بیماری Citrullinemia
فهیمه تیموری،اعظم شفائی،دکتر عبدالرضا وارسته – بخش بیماریهای متابولیک آزمایشگاه پردیس مشهد
دکتر عبدالرضا وارسته- بخش بیماریهای متابولیک آزمایشگاه پردیس مشهد،
و مركز آموزش عالي علوم پزشكي وارستگان مشهد ومركز تحقيقات ايمونولوژي مشهد
مقدمه:
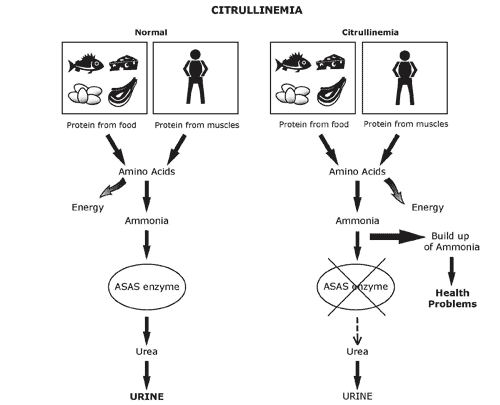
بیماری سیترولینمی یکی از اختلالات چرخه اوره میباشد. اختلالات چرخه اوره با افزایش آمونیاک خون همراه هستند. چرخه اوره از پنج آنزیم، یک کوفاکتور و در نهایت دو پروتئین انتقالدهنده تشکیل شده است. نقص در هر یک از چهار آنزیم (CPSI, OTC, ASS, ASL) موجود در این چرخه و همچنین نقص در کوفاکتور NAGS منجر به افزایش و تجمع آمونیاک در بدن میشود.
CPSI : کربامیل فسفات سنتتاز 1OTC : اورنیتین ترانس کربامیلاز
ASS : آرژنین سوکسینیک سنتتازASL : آرژنینوسوکسینک اسید لیاز
NAGS : N-استیل گلوتامات سنتتاز
بیماری سیترولینمی که توسط افزایش آمونیاک در خون مشخص میشود، به دلیل نقص در دو آنزیم آرژینوسوکسینیک اسید سنتتاز و آرژنینوسوکسینک اسید لیاز بوجود میآید. همانطور که در بالا اشاره شد و در شکل نیز مشخص است، هر دو آنزیم در چرخه اوره نقش دارند. وظیفه این آنزیمها، هضم اسیدهای آمینه آسپارتات و آرژینوسوکسینات و دفع آمونیاک بدن میباشد. نقص آنها با افزایش شدید آمونیاک و اسیدآمینه سیترولین در خون، همراه خواهد بود. افزایش آمونیاک خون در این بیماران، معمولاً به دنبال یک دوره گرسنگی طولانی، در هنگام عفونت و بیماری و یا بعد از دریافت مقدار زیادی پروتئین دیده میشود. افزایش سطح این مواد در خون، میتواند باعث ایجاد آسیبهای مغزی و حتی مرگ شود.
علائم بالینی:
بیماری سیترولینمی دوتیپ مختلف دارد. موتاسیون در ژن ASS1 منجر به ایجاد تیپ یک بیماری سیترولینمی (نقص آرژینوسوکسینیک اسید سنتتاز) میگردد. در این تیپ بیماری به دلیل نقص آرژینوسوکسینیک اسید سنتتاز، آرژینوسوکسینات در خون بیماران کاهش و سیترولین افزایش مییابد. علائم تیپ یک این بیماری که سیترولینمی کلاسیک نیز خوانده میشود، معمولاً در روزهای ابتدای تولد دیده میشود. نوزادان مبتلا، در ابتدای تولد سالم به نظر میرسند، ولی هنگامی که آمونیاک در خون این بیماران افزایش مییابد، معمولاً سستی و ضعف، اشکال در غذا خوردن، استفراغ و کاهش هوشیاری در بیمار دیده میشود. این مشکلات در بسیاری از موارد، زندگی نوزاد را تهدید میکنند. فرم خفیف تری از تیپ یک سیترولینمی ممکن است در دوران کودکی و بزرگسالی نیز بروز یابد. این فرم با شروع دیررس، با سردردهای شدید، کاهش نسبی بینایی، ضعف ماهیچهها، آتاکسی و بیحالی همراه میباشد. تعداد اندکی از افرادی که موتاسیونهای تیپ یک را دارند، هیچگاه علائمی از این بیماری را بروز نمیدهند.
موتاسیون در ژن ASL موجب ایجاد تیپ دو (نقص آرژینوسوکسینیک اسید لیاز) میشود. در این تیپ بیماری به دلیل نقص آرژینوسوکسینیک اسید لیاز، آرژینوسوکسینات و سیترولین در خون بیماران، افزایش مییابد. تیپ دو این بیماری به طور عمده سیستم عصبی را درگیر مینماید و باعث گیجی، بیقراری، از دست دادن حافظه، رفتارهای غیر طبیعی (تحریکپذیری بالا و بیش فعالی)، تشنج و کوما میشود. در بعضی از مبتلایان این علائم در بزرگسالی بروز مییابند. این علائم میتوانند تهدیدکننده زندگی فرد باشند. علائم تیپ دو سیترولینمی در بزرگسالان (CTLN2) معمولاً به دنبال یک عفونت، جراحی، یک درمان خاص یا دریافت میزان بالای الکل شروع میشوند.
تیپ دو بزرگسالان ممکن است در افرادی دیده شود که در دوران کودکی اختلال کبدی به نام کلستاز داخل کبدی نوزادی (NICCD) داشتهاند که این بیماری در نتیجه کمبود سیترین ایجاد میگردد. فرم NICCD، به عنوان تیپ دو سیترولینمی با شروع نوزادی نیز خوانده میشود.
|
موتاسیون در ژن ASS1 |
موتاسیون در ژن ASL |
||
|
سیترولینمی تیپ 1 (نقص آرژینوسوکسینیک اسید سنتتاز) |
سیترولینمی تیپ 2 (نقص آرژینوسوکسینیک اسید لیاز) |
||
|
فرم نوزادی
|
فرم با شروع دیررس
|
سیترولینمی تیپ 2 در بزرگسالان (CTLN2)
|
کلستاز داخل کبدی نوزادی (NICCD)
|
|
|
|
|
|
NICCD: Neonatal intrahepatic cholestasis caused by citrin deficiency
CTLN2: Adult-onset type II citrullinaemia
روش تشخیص:
غربالگري نوزادان به وسيله تكنيك TMS سطح بالايي از سیترولین را در هر دو تیپ بیماری سیترولینمی نشان ميدهد. مقدار سیترولین در نقص آرژنینوسوکسینیک اسید سنتتاز تا 100 برابر میزان طبیعی آن ميرسد. بيماران مبتلا به نقص آرژنینوسوکسینات لیاز، سطح قابل اندازهگيري از آرژینوسوکسینیک اسید در پلاسمای خود دارند كه در حالت عادي در پلاسما قابل اندازهگیری نيست. فعاليت هر دو آنزيم ميتواند از طريق بيوپسي كبد اندازهگیری شود. ژنهای درگیر در هر دو تیپ بیماری (ASS1 در تیپ یک وSLC25A13 در تیپ دو) و جهشهای آنها شناسايي شده است. در صورت حضور جهش در هر دو والدین، بررسی DNA براي تشخيص قبل از تولد ميتواند استفاده شود. مطالعات بيوشيميايي آمينوسيتها و پرزهاي كوريوني نیز میتواند کمک کننده باشد. علاوه بر آن در بیماران مبتلا به نقص آرژنینوسوکسینات لیاز، حضور آرژینوسوکسینیک اسید در مايع آمنيوتيك بيماران در تشخيص قبل از تولد بسيار مؤثر است.
درمان:
به نظر ميرسد که علائم بيماري سیترولینمی، بیشتر به دلیل افزایش آمونیاک باشد تا تجمع سیترولین. جهت درمان افزایش حاد آمونیاک خون، میتوان بیمار را همودیالیز کرد. تجویز بنزووات سديم منجر به کونژوگه شدن آن با گلایسین شده و باعث تسهیل دفع ادراری آن میشود. تزريق وريدي آرژنين منجر به پاكسازي آمونياك، به وسيله تشكيل سیترولین در هر دو نقص خواهد شد. هر دو اين متابوليتها (سیترولین و گلایسین) به وسیله ادرار از بدن دفع میشوند و نيتروژن اضافي را از آمونياك ميگيرند. بيماراني كه از دوران نوزادی نشانههاي بيماري را دارند، بايد رژیم غذایی خاصی را رعایت کنند. غذاهای طبی و شیر خشکهای مخصوص برای این نوزادان در دسترس میباشد. برای کودکان نیز رژیم غذایی کم پروتئین و دارای کربوهیدرات بالا توصیه میشود.
اغلب کودکانی که بعد از تولد سریعاً تحت درمان قرار میگیرند، زندگی عادی همراه با رشد و نمو و هوش طبیعی خواهند داشت. درمان فوری احتمال افزایش آمونیاک خون را کاهش میدهد. در برخی از بیماران، علیرغم درمان، باز هم حملات ناگهانی افزایش آمونیاک خون دیده میشود. در این کودکان آسیبهای مغزی، مشکلات یادگیری، عقبماندگی ذهنی و تشنج مشاهده میشود.
توارث:
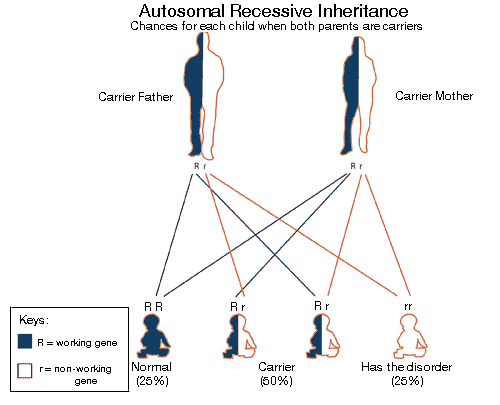
اين نقص معمولاً از الگوي وراثتي اتوزومال مغلوب پيروي ميكند. در اين الگو بيماران مبتلا باید دو كپي از ژن جهش يافته را براي بروز علائم داشته باشند. افرادي كه داراي يك ژن معیوب هستند ناقل خوانده ميشوند و علائم بیماری را نشان نميدهند، اما ميتوانند ژن معیوب را به فرزندان خود منتقل كنند. وقتی هر دو والدین ناقل باشند، 25% احتمال تولد کودک سالم، 50% احتمال تولد کودک ناقل و 25% احتمال تولد کودک مبتلا به سيترولينمي، در هر بارداری وجود دارد.
شیوع بیماری:
برخی منابع شیوع تیپ یک سیترولینمی را حدود 1 مورد در هر 57,000 تولد ذکر کردهاند. تیپ دو این بیماری که معمولاً در ژاپنیها دیده میشود، شیوع کمتری داشته و حدود 1 مورد در هر 100,000 تا 200,000 گزارش شده است، اما پیشبینی میشود شیوع این بیماری در کشورهایی همانند ایران که ازدواجهای فامیلی رواج بیشتری دارند، بیشتر باشد.
طبق بررسیهای انجام شده در آزمایشگاه پردیس مشهد از بین 1466 کودک که جهت بررسی بیماریهای متابولیک توسط تکنیک TMS غربالگری شده بودند، 4 نفر مبتلا به بیماری سیترولینمی شناسایی شدند. بدیهی است که این تعداد بیمار و روش بررسی آنها نمیتواند میزان شیوع این بیماری را در جامعه ما مشخص نماید، اما میتواند بیانگر شیوع احتمالی بالاتر این نقص، در جامعه ما نسبت به شیوع جهانی آن باشد. یقیناً استفاده از روشهای غربالگری پیشرفته میتواند تصویر دقیق تری از شیوع این بیماری را در جامعه ما ارائه نماید.
با توجه به اهمیت زمان در درمان و پیشگیری از عوارض سوء این بیماری و تحمیل هزینههای روحی و مالی این بیماری به خانواده و جامعه، منطقی به نظر میرسد که غربالگری این بیماری همانند اکثر کشورهای پیشرفته دنیا و اغلب کشورهای همسایه، در برنامه غربالگریهای روتین برای تمام نوزادان متولد شده در ایران قرار گیرد.
References:
1) Perkin Elmer Genetics, Newborn Screening
2) American College of Medical Genetics, 2006
3) http://www.ccmckids.org/research/
5) Medical Genetic Searches: A specialized PubMed search designed for clinicians that
is located on the PubMed Clinical Queries page.
6) http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=citrin
7) rare disease day 2010-citrullinemia
•9) Brusilow, S.W. and Horwich, A. Urea Cycle Enzymes. In, The Metabolic and Molecular Basis of Inherited Disease. 8th Edition, 2001. Scriver, Beaudet, et al. McGraw-Hill.
10) Maestri, N.E., Hauser, E.R., Bartholomew, D., et al. Prospective treatment of urea cycle disorders.
•11) Maestri, N.E., Clissold, D.B., Brusilow, S.W. Long-term survival of patients with Argininosuccinate Synthetase deficiencyRutledge, S.L.
12)Havens, P.L., Haymond, M.W., et al. Neonatal hemodialysis: Effective therapy for the encephalopathy
